
Strong Clinical Signal, Inconsistent Public Disclosure
Disitamab Vedotin plus Toripalimab in First-Line Urothelial Carcinoma
Executive Summary (Revised with Temporal Disclosure Inconsistency)
The Phase 3 randomized trial of disitamab vedotin combined with toripalimab in previously untreated HER2-expressing advanced or metastatic urothelial carcinoma, published in The New England Journal of Medicine, reports large and statistically robust efficacy outcomes compared with platinum-based chemotherapy.
In the intention-to-treat population, median progression-free survival reached 13.1 months in the experimental arm versus 6.5 months under chemotherapy, corresponding to a hazard ratio of approximately 0.36. Median overall survival was 31.5 months versus 16.9 months, with a hazard ratio of approximately 0.54. The objective response rate exceeded 70% in the disitamab vedotin–toripalimab arm compared with roughly 50% in the control group. Grade ≥3 treatment-related adverse events were reported less frequently in the experimental arm.
The published results are attributed to a single Phase 3 study identified as NCT05302284, which is the sole trial identifier referenced in the article.
According to the publicly accessible ClinicalTrials.gov record for NCT05302284, the study is listed as Recruiting/Active and reports two locations, both in Beijing. The registry does not indicate study completion and does not report results. In addition, the registry lists a Study Completion Date in 2028, which remains unchanged at the time of publication.
In contrast, corporate press releases issued by the sponsor (RemeGen Co., Ltd.) describe the same study as a completed multicenter Phase 3 trial conducted across 74 clinical sites in China, enrolling 484 patients, and achieving mature progression-free and overall survival outcomes.
No alternative urothelial carcinoma trial using the same disitamab vedotin–toripalimab combination is identifiable in ClinicalTrials.gov that could account for the published dataset under a different registration number. The only clearly visible Phase 3 program combining these agents outside this identifier is NCT05980481, conducted in gastric cancer.
Taken together, these facts establish documented inconsistencies across public disclosure channels—peer-reviewed publication, clinical trial registry, and corporate communications—regarding execution scale, study status, and temporal alignment of the trial underlying the reported results. Specifically, the publication of apparently mature overall survival data in 2025, alongside a registry-listed completion date of 2028, is not explicitly labeled as an interim analysis in the published article.
This analysis does not dispute the reported efficacy signal. It records a verifiable disclosure divergence that limits independent reconstruction of trial execution, timing, and data provenance based on publicly available records.
Analytical Framework (Formal Declaration)
This analysis applies BBIU’s Orthogonal Differentiation Protocol (ODP) and Differential Force Projection (DFP) framework, including the following indices:
ODP-Index™ — degree of structural revelation
DFP-Index™ — degree of external force projection
Composite Displacement Velocity (CDV) — tempo of structural exposure
Canonical Definitions
ODP (Orthogonal Differentiation Protocol):
A framework that reveals a system’s internal structure when independent forces — Mass (M), Charge (C), Vibration (V), and Inclination (I) — act orthogonally within the neutral medium of Time (T).
DFP (Differential Force Projection):
A framework that measures how much of a system’s internal force capacity is actually projected outward, conditional on cohesion (δ), structural coherence (Sc), and time.
Time is not a force.
It is the exposure medium.
Structural Diagnosis
1. Observable Surface (Pre-ODP Layer)
At the observable surface, the system presents as:
A Phase 3 oncology trial published in NEJM
Large hazard-ratio separations in both PFS and OS
A HER2-targeted ADC combined with PD-1 blockade
A Chinese sponsor with domestically approved biologics
No publicly disclosed FDA enforcement actions
Media and market narratives converge toward the interpretation of a globally practice-changing result, largely equating publication venue and numerical magnitude with universal validity.
This layer remains descriptive.
2. ODP Force Decomposition (Internal Structure)
2.1 Mass (M) — Structural Density
Structural mass is concentrated:
Clinical execution limited to two Beijing centers
Sponsor-controlled trial design and operations
Manufacturing and quality systems rooted in a single regulatory jurisdiction
This density increases internal coherence but reduces external adaptability.
2.2 Charge (C) — Polar Alignment
Internally, charge alignment is strongly positive: sponsor incentives, domestic regulatory priorities, and national innovation narratives reinforce one another.
Externally, polarity weakens. In FDA, EMA, and PMDA environments, the same alignment produces skepticism rather than attraction, as independence and heterogeneity are prioritized.
2.3 Vibration (V) — Resonance / Sensitivity
Surface volatility is low: clean datasets, smooth curves, and narrative consistency. This reflects damping through homogeneity, not proven robustness. Sensitivity to external audit or independent replication remains untested.
2.4 Inclination (I) — Environmental Gradient
The regulatory gradient is asymmetric:
Favorable slope toward domestic approval and reimbursement
Steep uphill gradient toward Western regulatory scrutiny
The system has not yet traversed this slope.
2.5 Temporal Flow (T)
Time currently benefits the system. Publication and narrative consolidation precede full regulatory stress testing, allowing confidence to accumulate ahead of verification.
ODP-Index™ Assessment — Structural Revelation
The system’s internal structure is increasingly legible under pressure. Dominant forces are Mass and Charge, with external Inclination beginning to exert counter-pressure. Exposure is accelerating, not stabilizing.
The ODP-Index™ is therefore moderate to high, indicating partial but incomplete revelation.
Composite Displacement Velocity (CDV)
CDV is moderate and rising. Structural exposure is occurring gradually as global interpretation expands, but has not yet reached a regime-shift threshold.
DFP-Index™ Assessment — Force Projection
Internal Projection Potential (IPP) is evident: the drug demonstrates biological activity and clinical effect. However, cohesion (δ) and structural coherence (Sc) degrade across borders due to limited independent verification and untested manufacturing portability.
The system contains force, but cannot yet project it reliably into external regulatory environments. The DFP-Index™ is moderate internally and low externally.
ODP–DFP Interaction & Phase Diagnosis
The system occupies a High ODP / Low DFP phase: structural exposure without commensurate projection. This corresponds to an exposed non-agent state, where signal exists but remains jurisdiction-bound.
Trajectory, not snapshot, defines the risk.
Five Laws of Epistemic Integrity (Audit Layer)
Truth: Efficacy signal is real; global trust is unproven.
Reference: Anchoring relies on publication authority over regulatory stress.
Accuracy: Mechanisms are correctly described; execution limits are underweighted.
Judgment: Effect size is overweighted relative to verification depth.
Inference: Global conclusions exceed structural constraints.
BBIU Structural Judgment
The system is generating a strong domestic signal that is being over-extended in interpretation. The deferred adjustment is independent verification, both clinical and manufacturing. Current responses rely on authority substitution rather than structural reinforcement, and therefore cannot resolve the underlying ODP.
BBIU Opinion (Controlled Interpretive Layer)
Structural Meaning:
The case illustrates how high-magnitude results from homogeneous environments can dominate discourse before portability is established.
Epistemic Risk:
Conflating publication and effect size with global readiness increases downstream correction costs when verification is imposed.
Comparative Framing:
Unlike globally distributed trials underpinning other ADC-ICI combinations, this program remains geographically concentrated.
Strategic Implication (Non-Prescriptive):
Signal discovery has outpaced verification capacity.
Forward Structural Scenarios (Non-Tactical)
Continuation under current conditions preserves narrative stability while exposure accumulates. A forced adjustment emerges when bridge trials, independent adjudication, or manufacturing inspections are required. An external shock—clinical attenuation or CMC deficiency—would sharply increase CDV.
Why This Matters (Institutional Lens)
For regulators, it reinforces the distinction between efficacy and verifiability.
For institutions, it clarifies the gap between directional validity and asset portability.
For long-horizon capital, it highlights the cost of premature consensus.
For strategic actors, it demonstrates how jurisdiction-bound trust constrains projection.
Access & Scope Note
Extended diagnostics, index trajectories, and ODP–DFP phase mapping are available under BBIU Institutional Access.
References
Peer-Reviewed Primary Source
The New England Journal of Medicine — Disitamab Vedotin plus Toripalimab in HER2-Expressing Advanced Urothelial Cancer (NEJMoa2511648). New England Journal of Medicine
Trial Registry
ClinicalTrials.gov — NCT05302284 (A Study of RC48-ADC Combined With Toripalimab…). ClinicalTrials
Sponsor Press Releases
PR Newswire (RemeGen) — May 12, 2025: RC48-C016 Phase 3 reached dual primary endpoints (PFS/OS). PR Newswire
PR Newswire (RemeGen) — Oct 20, 2025: ESMO 2025 Presidential Symposium communication on RC48-C016 (study description and positioning). PR Newswire
RemeGen official website (Company News mirror) — Oct 20, 2025: RC48-C016 post (includes enrollment/center claims as stated by sponsor). remegen.com+1
Congress / Presentation Record
ESMO Congress 2025 (OncologyPRO) — presentation page for DV + toripalimab vs chemotherapy in 1L la/mUC with HER2 expression. Oncology Pro
Independent Secondary Summaries (Context / Cross-checking)
Annex 1 – Integrated Pharmacological and Pharmacokinetic Characterization of Disitamab Vedotin, Toripalimab, and Monomethyl Auristatin E (MMAE)
(Technical Annex – Mechanistic, Pharmacological, and PK Reference)
1. Disitamab Vedotin (RC48, Aidexi®)
1.1 Molecular Composition and ADC Architecture
Disitamab vedotin is a HER2-directed antibody–drug conjugate (ADC) composed of three integrated elements:
A humanized IgG1 monoclonal antibody directed against the extracellular domain of HER2
A protease-cleavable linker system
The cytotoxic payload monomethyl auristatin E (MMAE)
The antibody backbone is engineered to maintain high-affinity binding across a broad HER2 expression spectrum, including tumors with low antigen density (IHC 1+). The conjugate exhibits a relatively elevated drug–antibody ratio (DAR) compared with early-generation HER2 ADCs, increasing intracellular payload delivery per internalization event while simultaneously increasing sensitivity to manufacturing variability.
1.2 Functional Role of HER2: Delivery Marker Rather Than Driver Dependency
Within disitamab vedotin, HER2 functions primarily as a cell-surface delivery address, not as an obligate oncogenic driver. Therapeutic activity does not depend on sustained inhibition of HER2 signaling pathways or on gene amplification.
Clinical implications include:
Activity in tumors lacking HER2 amplification
Dependence on antigen-mediated internalization rather than pathway addiction
Reduced relevance of HER2 signaling resistance mechanisms
HER2 defines where the payload is delivered, not why the tumor is vulnerable.
1.3 Mechanism of Action
The pharmacological sequence proceeds as follows:
Binding of the ADC to HER2 on the tumor cell surface
Receptor-mediated endocytosis of the ADC–HER2 complex
Lysosomal trafficking and linker cleavage
Release of free MMAE into the cytoplasm
Inhibition of tubulin polymerization, leading to G2/M cell-cycle arrest
Activation of intrinsic apoptotic pathways
Following apoptosis, loss of membrane integrity allows MMAE to diffuse into adjacent tumor cells, generating a localized bystander cytotoxic effect independent of HER2 expression.
1.4 Pharmacokinetics of Disitamab Vedotin (ADC-Level PK)
The pharmacokinetics of disitamab vedotin are dominated by the monoclonal antibody component, with payload exposure determined secondarily by linker stability and intracellular processing.
Administration
Intravenous infusion
Distribution
Predominantly intravascular
Tumor accumulation mediated by HER2 binding
Limited penetration into immune-privileged compartments, including the central nervous system
Plasma Behavior
Biphasic concentration–time profile typical of IgG-based ADCs
Initial distribution phase followed by prolonged elimination
Circulating ADC remains largely intact under conditions of adequate linker stability
Clearance
Proteolytic degradation via the reticuloendothelial system
Not primarily renal
Influenced by target-mediated drug disposition (TMDD), particularly in settings of high tumor HER2 burden
Half-life
Measured in days
Shortened by high antigen sink, anti-drug antibodies, or accelerated clearance mechanisms
ADC-level PK determines exposure duration, tumor delivery efficiency, and systemic safety margin.
1.5 Safety Profile
Observed toxicities are predominantly payload-driven rather than HER2-mediated and include:
Peripheral sensory neuropathy
Myelosuppression
Gastrointestinal toxicity
Fatigue and asthenia
HER2-associated cardiotoxicity is less prominent than with unconjugated HER2 antibodies, reflecting the delivery-centric mechanism.
1.6 Manufacturing and CMC Sensitivities
Disitamab vedotin exhibits high CMC sensitivity, particularly in:
Consistency of drug–antibody ratio
Control of unconjugated (free) MMAE
Linker stability during storage and transport
Prevention of cross-contamination in cytotoxic handling suites
Cleaning validation for highly potent payload residues
CMC robustness is a primary determinant of regulatory portability.
2. Monomethyl Auristatin E (MMAE)
2.1 Chemical and Pharmacological Class
MMAE is a synthetic antineoplastic agent derived from dolastatin-10, belonging to the class of microtubule polymerization inhibitors. Its intrinsic cytotoxic potency lies in the sub-nanomolar range, rendering it unsuitable for systemic administration outside targeted delivery systems.
2.2 Mechanism of Action
MMAE binds with high affinity to β-tubulin, near the vinca alkaloid binding site, resulting in:
Inhibition of tubulin polymerization
Mitotic spindle collapse
G2/M cell-cycle arrest
Activation of intrinsic apoptotic pathways
The effect is strongest in proliferating cells but may affect non-dividing cells at sufficient intracellular concentrations.
2.3 Cellular Uptake and Bystander Effect
MMAE is:
Low molecular weight
Highly lipophilic
Readily membrane permeable
Once released intracellularly, MMAE distributes rapidly within the cytosol. Following apoptosis of the target cell, extracellular MMAE may diffuse passively into neighboring cells without receptor-mediated endocytosis, producing a local bystander effect, including in antigen-negative tumor cells.
This property enhances efficacy in heterogeneous tumors but also contributes to off-target toxicity.
2.4 Pharmacokinetics of MMAE (Payload-Level PK)
MMAE does not possess a clinically acceptable standalone pharmacokinetic profile. All relevant PK considerations arise after release from the ADC.
Release Context
Predominantly intracellular via lysosomal cleavage
Secondary extracellular release following apoptosis or necrosis
Distribution
Rapid intracellular diffusion
Local interstitial spread
Limited systemic redistribution under stable ADC conditions
Plasma Exposure
Low and transient when linker integrity is preserved
Increased systemic exposure correlates with toxicity risk
Metabolism
Primarily hepatic
Mediated by CYP3A4/5
Elimination
Predominantly biliary–fecal
Minimal renal clearance
Half-life
Very short when free
Clinically relevant exposure depends on ADC cleavage rate, tumor burden, and cumulative dosing
Drug–Drug Interaction Potential
Increased exposure with strong CYP3A inhibitors
Reduced exposure with strong CYP3A inducers
2.5 Toxicological Profile
MMAE-associated toxicities include:
Peripheral axonal neuropathy
Bone marrow suppression
Gastrointestinal mucosal injury
Fatigue
These toxicities are mechanism-intrinsic and independent of antigen expression.
2.6 Resistance Mechanisms
MMAE is a substrate for P-glycoprotein (ABCB1/MDR1). Upregulation of efflux transporters may reduce intracellular exposure and contribute to acquired resistance.
3. Toripalimab (JS001)
3.1 Molecular Classification
Toripalimab is a humanized IgG4 monoclonal antibody targeting programmed death-1 (PD-1). The IgG4 backbone minimizes Fc-mediated effector functions.
3.2 Mechanism of Action
Toripalimab blocks PD-1 interaction with PD-L1 and PD-L2, resulting in:
Restoration of T-cell activation
Increased cytokine production
Enhanced cytotoxic immune responses within the tumor microenvironment
It exerts no direct cytotoxic effect on tumor cells.
3.3 Pharmacokinetics of Toripalimab
Administration
Intravenous infusion
Distribution
Systemic, including lymphoid tissues
Limited CNS penetration
Plasma Behavior
Linear pharmacokinetics across therapeutic dose ranges
No clinically significant target-mediated saturation
Clearance
Reticuloendothelial catabolism
Not metabolized via CYP enzymes
Not renally cleared
Half-life
Consistent with IgG monoclonal antibodies (weeks)
Drug–Drug Interactions
No meaningful CYP-mediated interactions
PK unaffected by concomitant ADC administration
3.4 Safety Profile
Toxicities reflect immune activation and include:
Dermatologic reactions
Endocrinopathies
Immune-mediated colitis
Pneumonitis
Severity ranges from mild to life-threatening.
4. Integrated Pharmacological and PK Interaction Profile
There is no direct pharmacokinetic interaction between disitamab vedotin, MMAE, and toripalimab. Each component follows a distinct disposition pathway.
Functional coupling occurs at the pharmacodynamic level, where ADC-mediated tumor cell death enhances antigen release and immune priming, while PD-1 blockade sustains immune-mediated control.
5. Mechanistic Rationale for Combination: Cytotoxic Ceiling and Immune Sustainment
Disitamab vedotin demonstrates clear cytotoxic activity as a monotherapy through targeted delivery of MMAE, resulting in rapid tumor debulking and objective responses across HER2-expressing tumors, including those with low antigen density. However, this activity is intrinsically self-limiting.
The limitation does not arise from insufficient potency, but from structural constraints inherent to MMAE-based cytotoxicity:
Cytotoxic efficacy depends on proliferative status and intracellular payload accumulation
Dose escalation is constrained by cumulative, payload-driven toxicities (notably peripheral neuropathy and myelosuppression)
Selective pressure favors the emergence of resistant or slow-cycling tumor clones
Tumor cell death alone does not generate durable immune memory
As a result, disitamab vedotin monotherapy typically achieves tumor control without consolidation, leading to response attenuation over time despite initial efficacy.
ADC-mediated microtubule disruption and apoptosis increase antigen availability through cellular fragmentation and release of damage-associated molecular patterns (DAMPs), thereby enhancing the probability of antigen cross-presentation by antigen-presenting cells. This process raises immune visibility of the tumor but does not, by itself, ensure sustained adaptive immune engagement. Activated T cells rapidly upregulate inhibitory checkpoints, particularly PD-1, entering functional exhaustion in the absence of checkpoint blockade.
Toripalimab addresses this immunological bottleneck, not by increasing tumor cell apoptosis directly, but by preventing premature immune disengagement. PD-1 blockade sustains effector T-cell activity in the context of antigen exposure generated by ADC-induced tumor injury, extending tumor control beyond the cytotoxic window defined by MMAE exposure.
This interaction represents a conditional and asymmetric synergy:
Disitamab vedotin initiates tumor damage and antigen release
Toripalimab preserves and prolongs immune pressure generated in response to that damage
Importantly, this synergy does not eliminate collateral payload exposure. MMAE released into the tumor microenvironment may diffuse into surrounding cells, including activated T cells and macrophages, creating a tension between enhanced antigen visibility and localized immune collateral damage. The clinical benefit of the combination therefore depends on maintaining a net balance in which immune sustainment outweighs payload-induced immune attrition.
The rationale for combination therapy is thus not insufficiency of disitamab vedotin efficacy per se, but the need to convert cytotoxic response into durable disease control without exceeding the narrow toxicity ceiling imposed by MMAE.
6. Off-Target Toxicity and Non-Target Organ Involvement
Although disitamab vedotin is designed to deliver its cytotoxic payload selectively to HER2-expressing tumor cells, the emergence of adverse events in non-target organs is an expected and mechanistically explainable outcome of the platform.
This phenomenon arises through two distinct but convergent pathways.
6.1 On-Target, Off-Tumor Exposure (HER2 Basal Expression)
HER2 is physiologically expressed at low levels in multiple normal tissues, including gastrointestinal epithelium, bronchial epithelium, renal tubules, skin, and cardiomyocytes. While antigen density and internalization rates in these tissues are substantially lower than in HER2-overexpressing tumors, they are not null.
As a consequence, a fraction of circulating disitamab vedotin may undergo limited internalization in normal HER2-expressing tissues, leading to localized payload release. This mechanism contributes primarily to low-to-moderate grade toxicities, such as gastrointestinal symptoms, fatigue, and mild hepatic enzyme elevations, but does not account for the dominant dose-limiting toxicities observed clinically.
6.2 Off-Target Payload Exposure (Dominant Toxicity Driver)
The principal source of clinically relevant adverse events is payload-driven off-target exposure following MMAE release.
Once liberated from the ADC—either intracellularly during tumor cell apoptosis or extracellularly through secondary diffusion—MMAE:
Loses any dependence on HER2 expression
Enters surrounding cells by passive diffusion
Exerts cytotoxic effects based on cellular vulnerability, not antigen specificity
This mechanism preferentially affects tissues characterized by high proliferative activity or critical dependence on intact microtubule dynamics.
6.3 Organs Most Commonly Affected
As a result, adverse events are most frequently observed in non-target organs, including:
Peripheral nervous system
Sensory neurons rely on intact microtubule-based axonal transport. MMAE-induced microtubule destabilization leads to cumulative peripheral neuropathy, the primary dose-limiting toxicity.Bone marrow
Hematopoietic progenitor cells are highly proliferative and vulnerable to antimitotic agents, resulting in neutropenia, anemia, and, less frequently, thrombocytopenia.Gastrointestinal epithelium
High cellular turnover predisposes to nausea, diarrhea, and mucosal injury.Local immune compartment
Activated T cells and macrophages within the tumor microenvironment may be exposed to diffusing MMAE, leading to functional impairment or apoptosis. This represents a form of immune collateral damage, distinct from immune-related adverse events associated with PD-1 blockade.
These toxicities occur independently of HER2 expression and reflect the intrinsic pharmacology of MMAE.
6.4 Determinants of Toxicity Magnitude
The extent of off-target toxicity is modulated by:
Local MMAE concentration gradients
Payload clearance and diffusion kinetics
Cellular proliferative state
Expression of efflux transporters (e.g., P-glycoprotein)
Importantly, toxicity does not scale linearly with dose escalation; instead, it increases disproportionately once payload exposure exceeds a narrow therapeutic window.
6.5 Structural Implication
The occurrence of adverse events in non-target organs confirms that the therapeutic ceiling of disitamab vedotin is defined by payload biology rather than target biology. HER2 determines delivery efficiency, but MMAE determines systemic risk.
This structural constraint explains why clinical development prioritizes combination strategies to extend durability of response rather than simple dose intensification.
Image: SHA256 791892AC97E34300D75C4805AE2100A702F7AB6FA05E238B18E842371513A3FC
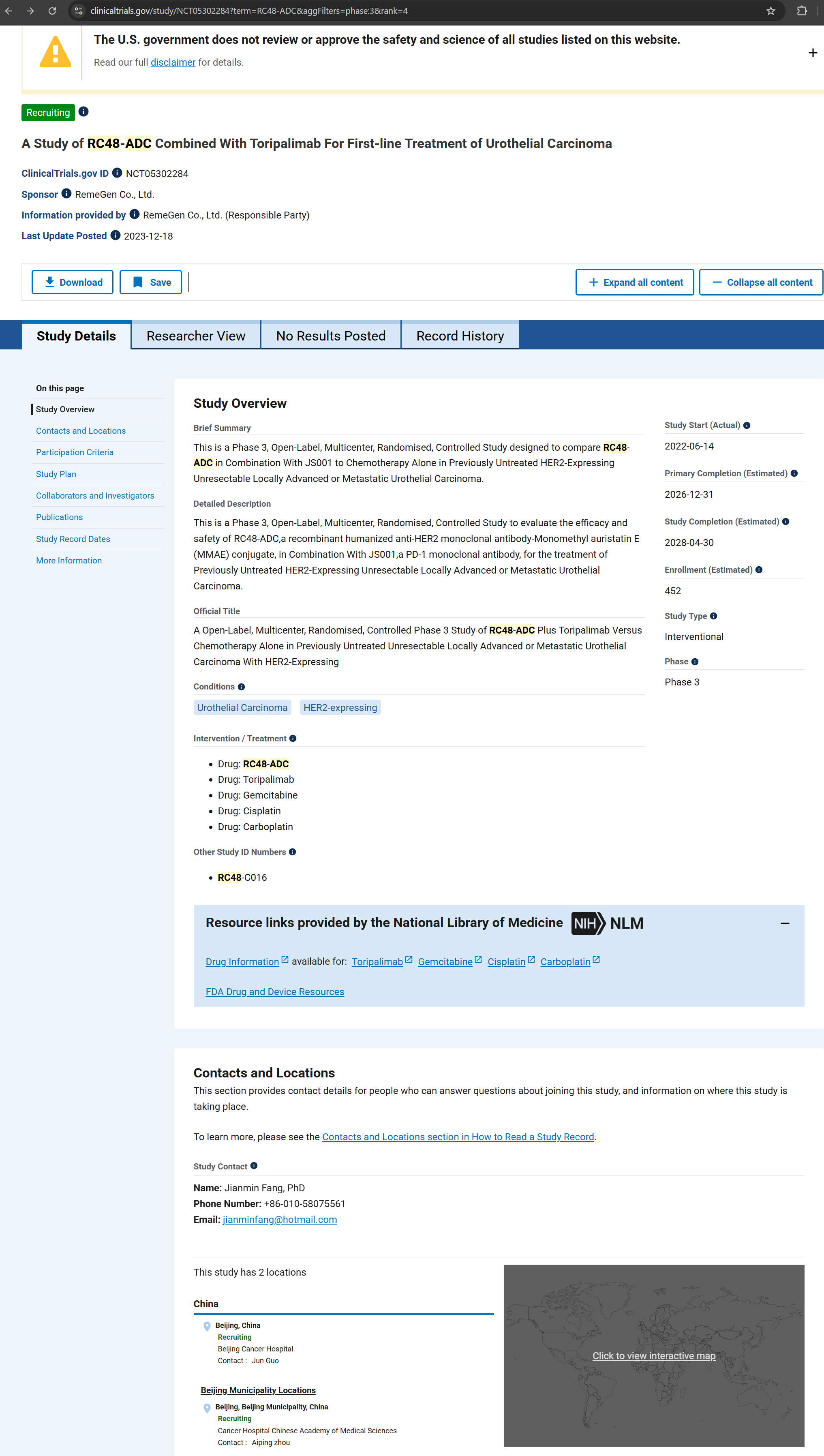
Image: SHA256 D5DF3A04BE1F61170B900234E99A6FDF3A4FC7420476D184A2BE5FDE7278FF75

Screenshots were archived with cryptographic hashes (SHA-256) to preserve integrity of public disclosures at the time of analysis.