
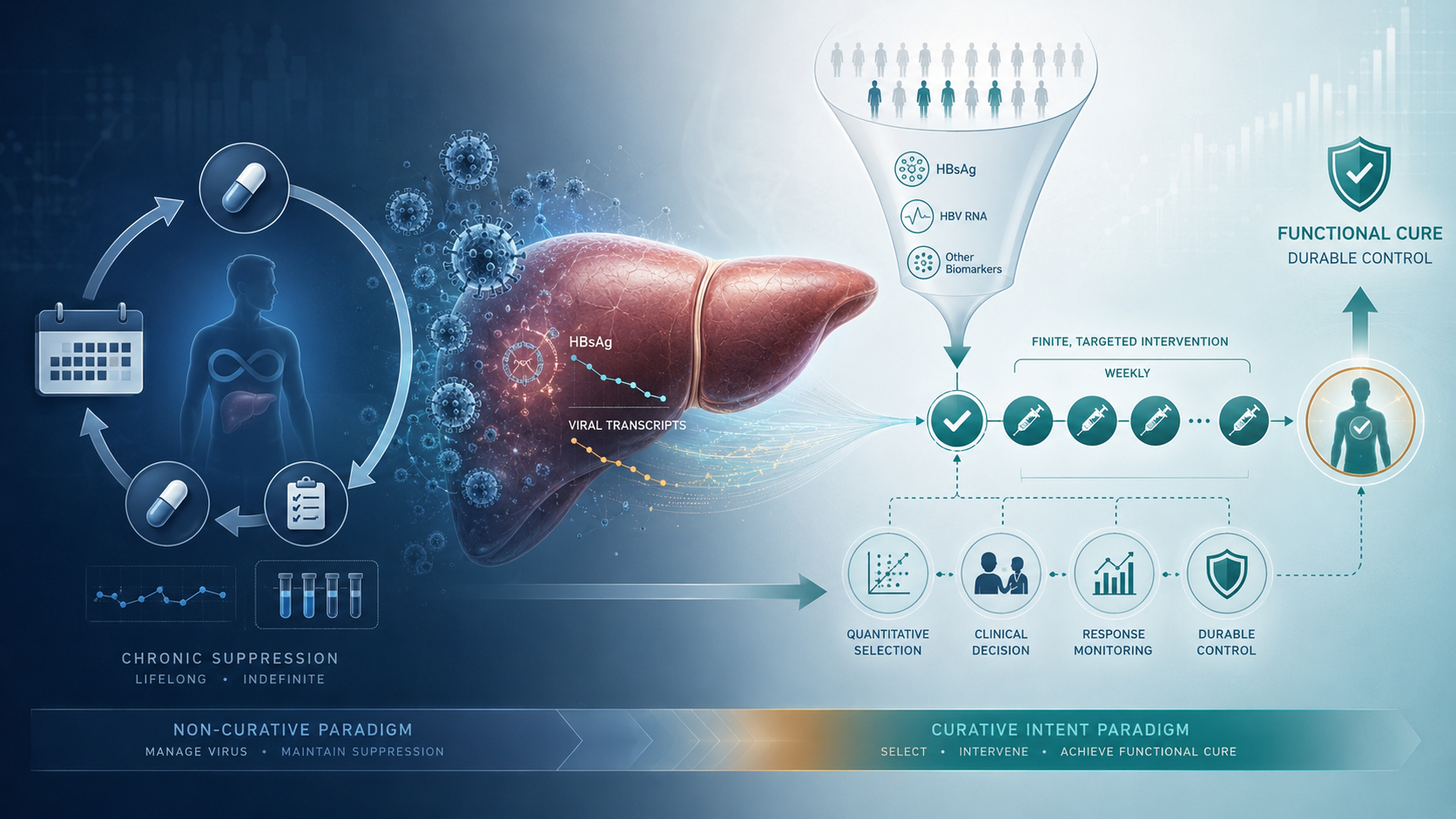
Bepirovirsen and the Structural Shift Behind Chronic Hepatitis B Treatment
Bepirovirsen’s Phase 3 data may signal more than a new treatment option for chronic hepatitis B. The deeper issue is that it could shift HBV management from indefinite viral suppression toward a biomarker-gated, finite intervention aimed at functional cure in selected patients.
This public analysis explains why the result matters beyond efficacy alone. Bepirovirsen may require health systems to rethink patient selection, quantitative HBsAg testing, treatment monitoring, ALT-flare interpretation, payer evaluation, and post-market evidence. Its potential is not that it replaces current HBV therapy for all patients, but that it may create a new treatment category for those who can move from chronic suppression to durable off-treatment control.
The institutional version expands this analysis with deeper assessment of the development program, pharmacology, dosing logic, health-economic burden, regional adoption constraints, payer exposure, and real-world safety uncertainty.

FDA’s First Generic Baloxavir Approval and the Market-Access Tension Behind Single-Dose Influenza Treatment
The FDA approval of the first generic version of Xofluza should not be read as a routine generic event. Baloxavir already existed as a branded single-dose antiviral; the new question is whether that convenience can move into a broader, payer-adoptable, seasonally scalable access category.
The approval may expand access, but it does not automatically resolve launch timing, pricing, payer behavior, clinical adoption, or stewardship concerns. Generic baloxavir still has to compete against inexpensive, familiar, and widely available oseltamivir, while proving that single-dose administration can create practical value within influenza’s narrow treatment window.
The strategic issue is therefore not only that FDA approved a generic product. The deeper issue is whether regulatory clearance can become real-world availability, reimbursement, early use, and measurable reduction in the clinical and economic burden of an influenza episode.
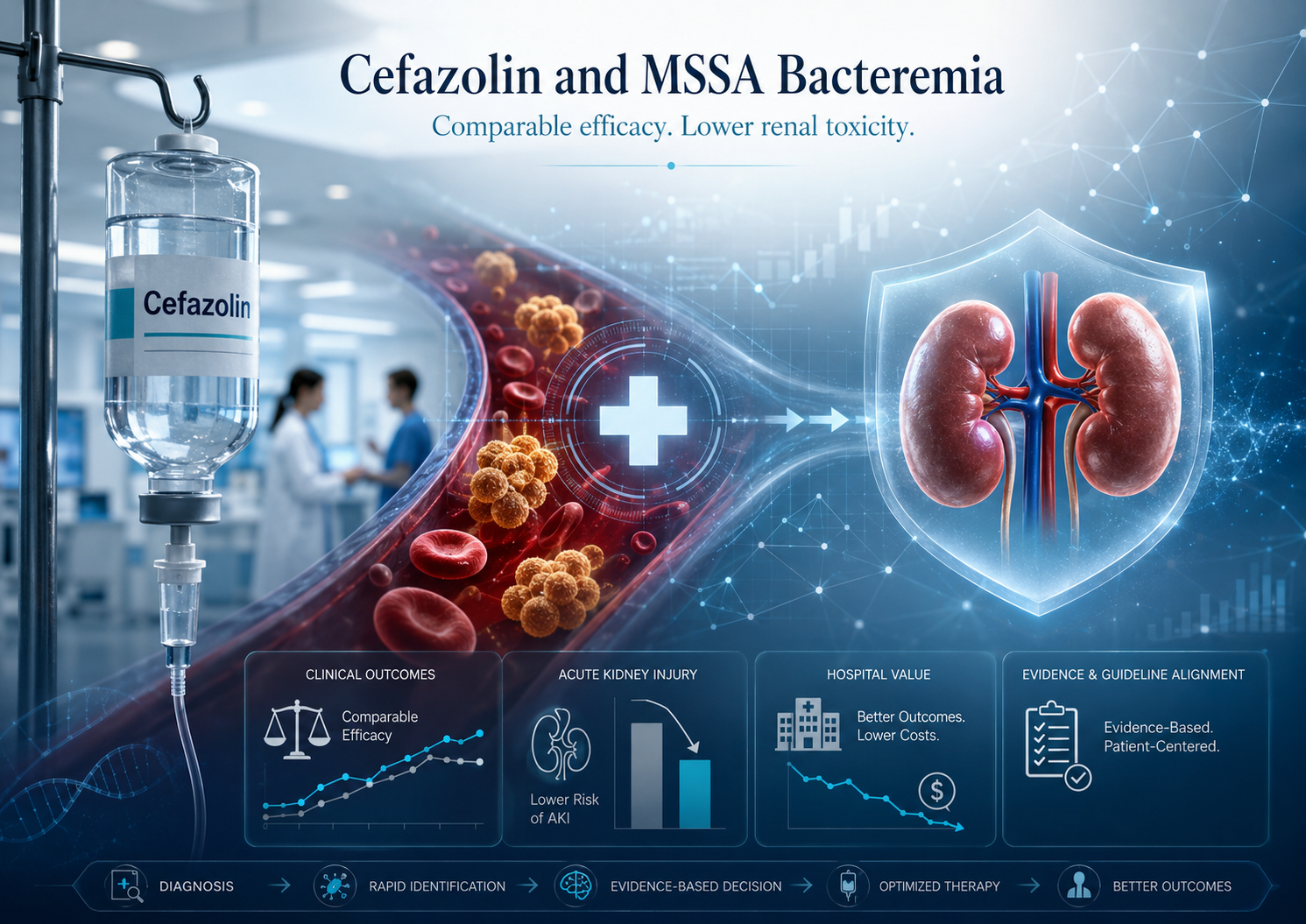
Cefazolin and the Structural Repricing of MSSA Bacteremia Treatment
A new NEJM/SNAP analysis suggests that cefazolin may reshape how hospitals think about MSSA bacteremia treatment. The finding is not about a new antibiotic, but about whether an established therapy can preserve comparable outcomes while reducing renal toxicity. The deeper question is institutional: when evidence shifts the balance between efficacy, safety, workflow, and patient burden, should legacy treatment defaults remain unchanged?
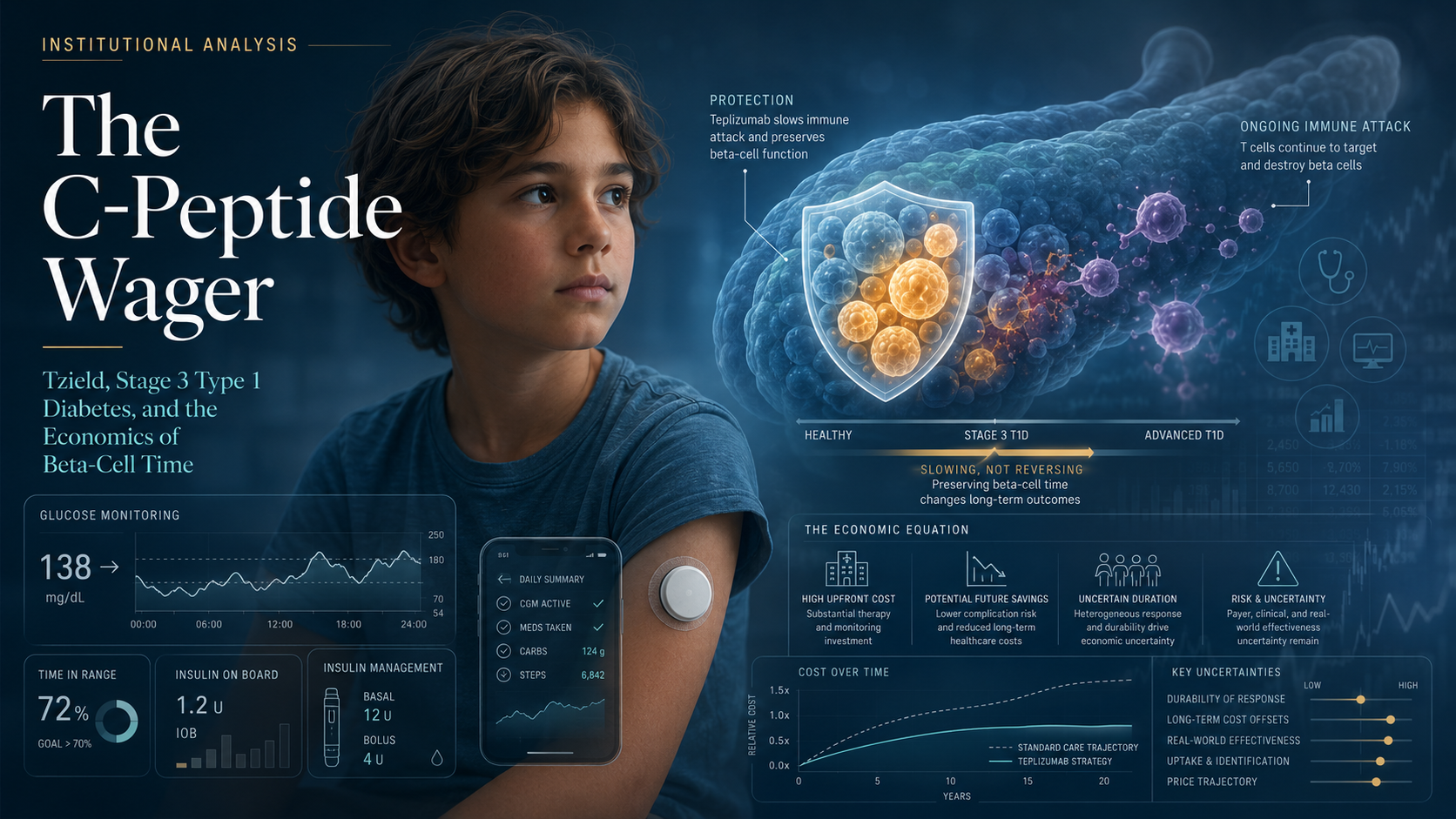
FDA’s Tzield Expansion and the Unresolved Value of Beta-Cell Time
FDA’s expanded approval of Tzield marks a regulatory shift in type 1 diabetes: beta-cell preservation is now an approvable therapeutic target, even after clinical diagnosis.
But the approval should not be mistaken for cure, disease reversal, or replacement of insulin-based care. In pediatric Stage 3 type 1 diabetes, the patient already has clinical disease and generally already requires insulin.
The unresolved question is whether preserving C-peptide can translate into durable clinical and economic value — or whether Tzield remains a scientifically meaningful but economically uncertain add-on to an already complex lifelong treatment model.

Dexcom Stelo’s Pediatric OTC Clearance and the Shift Toward Home-Based Metabolic Data
FDA’s pediatric OTC clearance of Dexcom’s Stelo is not only a device-access milestone. It signals a broader shift in continuous glucose monitoring: from prescription-based diabetes management toward home-based metabolic data infrastructure. As glucose biosensing moves into families, apps, and consumer health environments, the strategic question is no longer only whether glucose can be measured continuously, but whether that data can be used safely, responsibly, and transparently outside traditional clinical supervision.
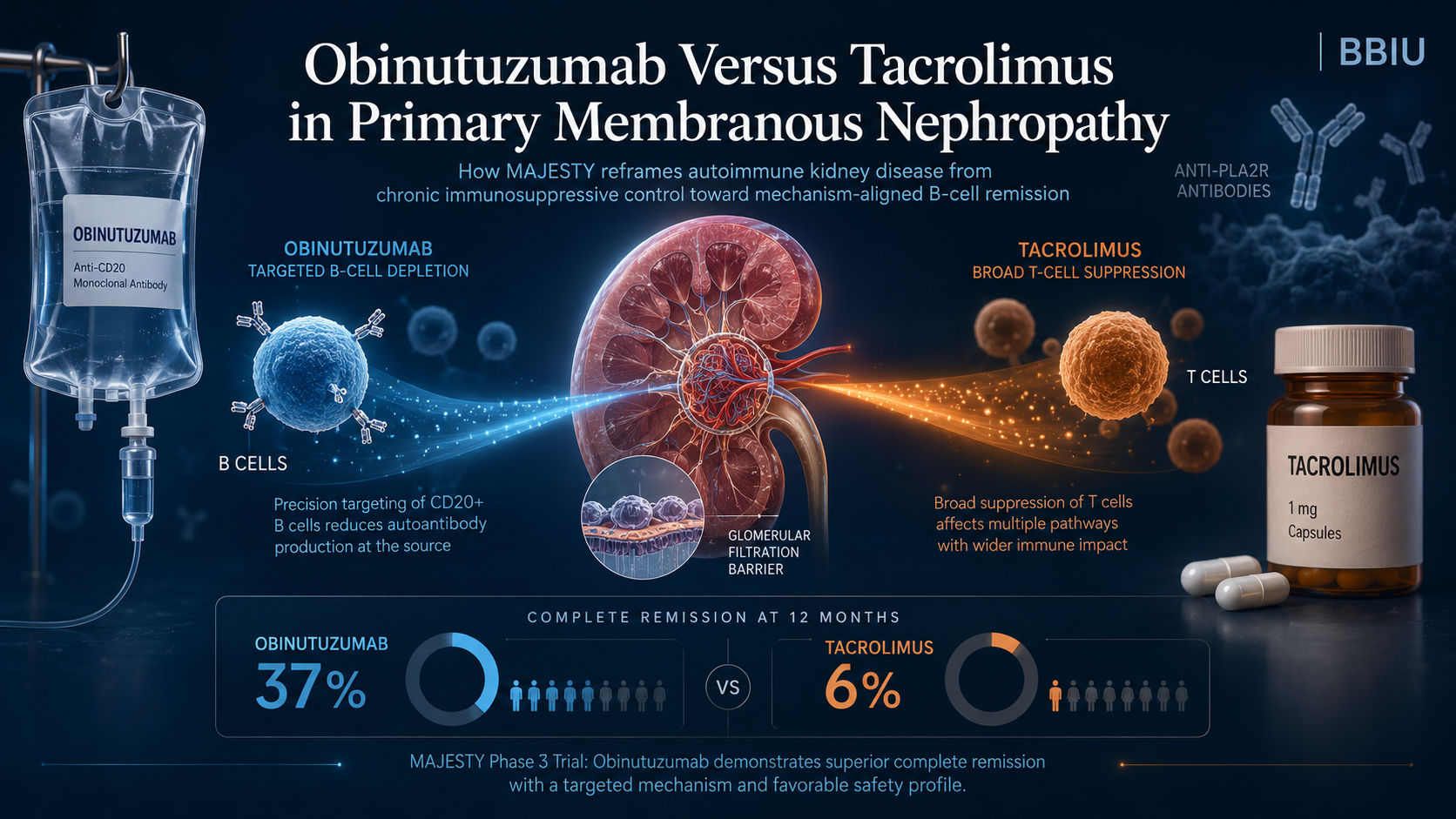
Obinutuzumab Versus Tacrolimus and the Reframing of Autoimmune Kidney Disease
MAJESTY does not simply compare obinutuzumab with tacrolimus. It compares two treatment logics in primary membranous nephropathy: targeted B-cell depletion versus chronic T-cell–oriented immunosuppression. The public result is clear — obinutuzumab achieved higher complete remission at Week 104 — but the deeper institutional question remains open: whether this remission signal can translate into durable immune control, stronger anti-PLA2R clearance, lower retreatment burden, and a new evidence standard for autoimmune kidney disease.
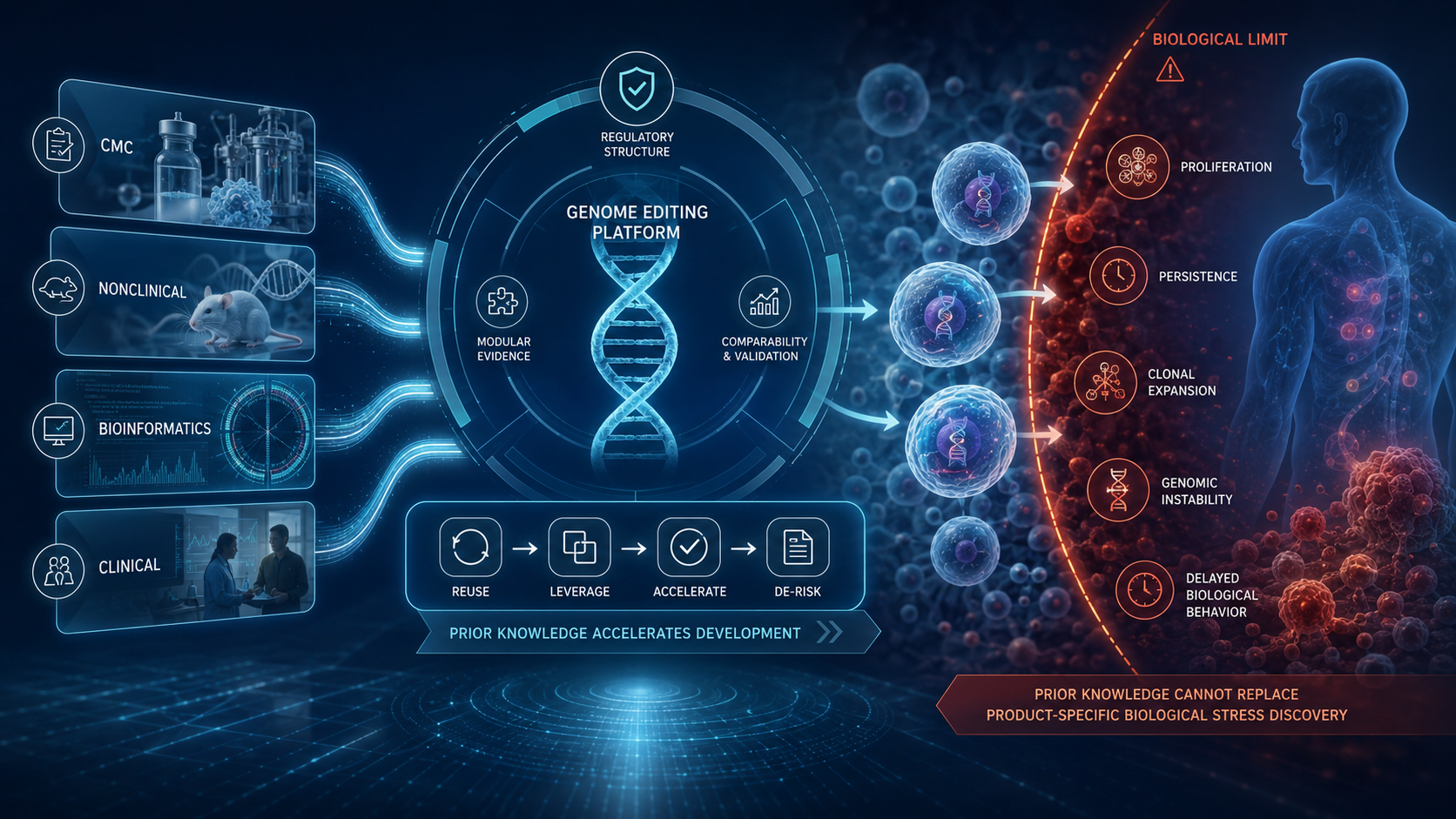
FDA’s Prior Knowledge Guidance and the Structural Repricing of Evidence in Genome Editing
FDA’s June 2026 draft guidance on leveraging prior knowledge in genome editing therapies is not simply an acceleration measure. It signals a structural shift in how evidence may be reused across gene therapy programs, provided that scientific similarity, applicability, and product-specific risk boundaries are clearly justified.
The deeper issue is not whether prior knowledge can reduce duplication. It is where prior knowledge reaches its biological limit.
For genome editing and gene-modified cellular therapies, reusable evidence may support CMC, nonclinical strategy, bioinformatics pipelines, and clinical planning. But it cannot replace product-specific interrogation of risks that emerge only after proliferation, persistence, clonal selection, genomic instability, or interaction with the patient’s biological environment.
BBIU’s public analysis identifies this boundary: regulatory acceleration is defensible only when it does not transfer foreseeable biological uncertainty from the sponsor’s development system into the patient’s body.

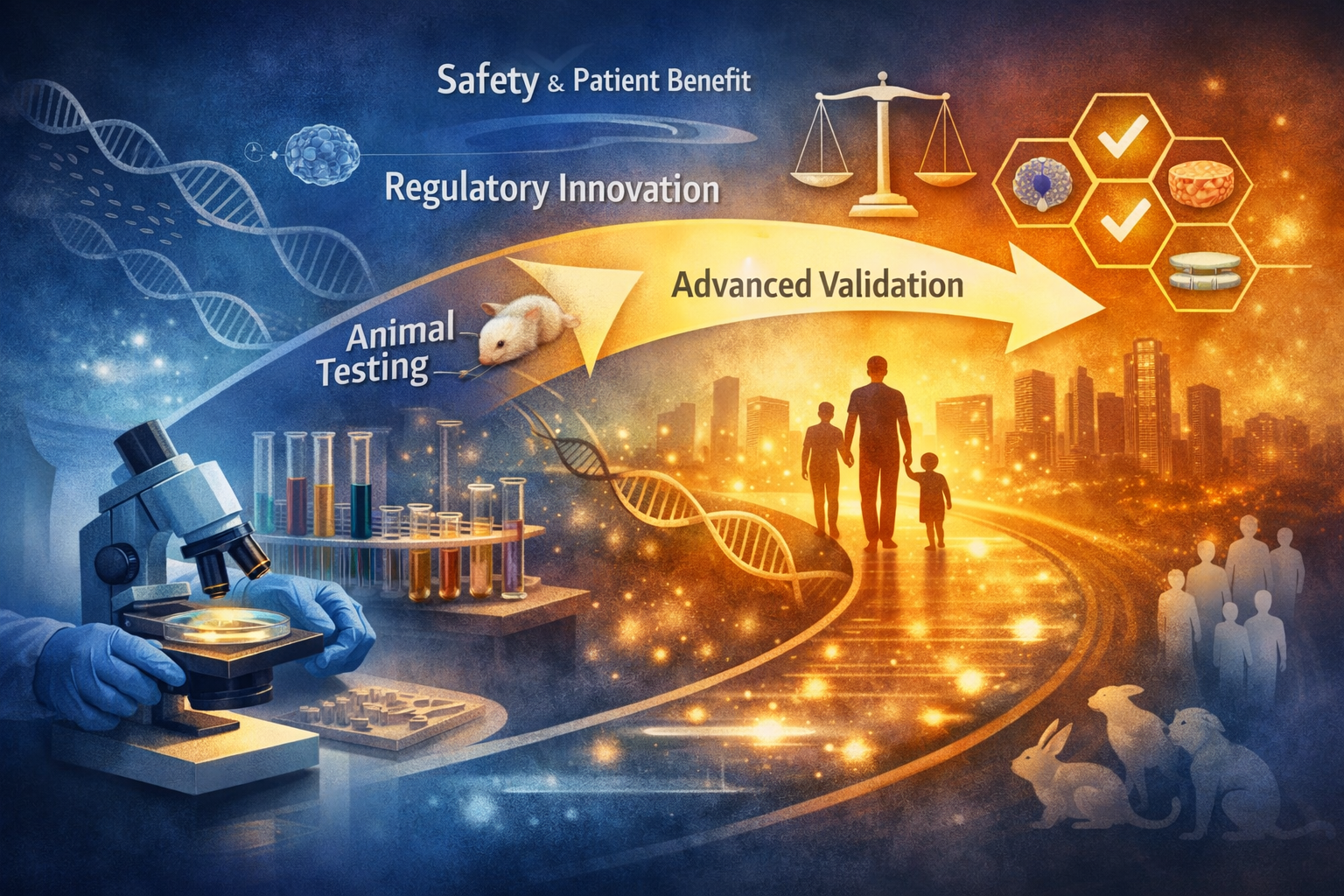
FDA’s Draft Guidance and the Repricing of Nonclinical Evidence in Oncology
FDA’s draft guidance on streamlined nonclinical safety studies in oncology should not be read as a broad elimination of animal testing. Its deeper significance is the redistribution of evidentiary responsibility. For selected oncology biologics and conjugated products, the regulatory value of an animal study will increasingly depend on whether the model is pharmacologically relevant, scientifically informative, and necessary to characterize human risk.
The shift is not from animal testing to no animal testing. It is from study volume to evidentiary logic. A smaller nonclinical package may be stronger if supported by target-binding data, pharmacological relevance, weight-of-evidence reasoning, and fit-for-purpose alternative methods. A larger package may be weaker if it relies on animal models that are procedurally familiar but biologically uninformative.
For sponsors, CROs, Regulatory Affairs teams, and investors, the central question becomes simple but decisive: is the evidence package sufficient because it is smaller, or because it is better?
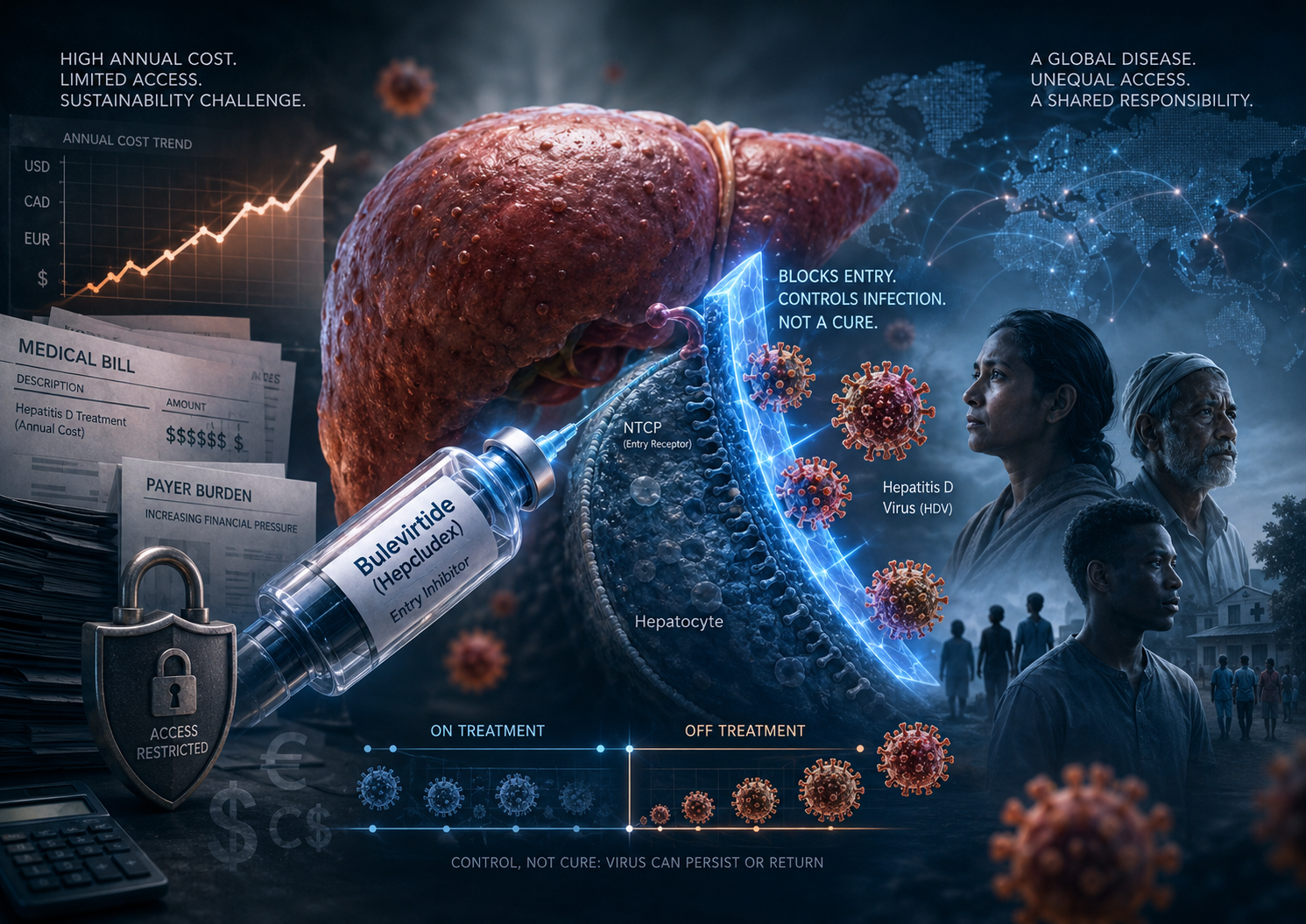
Hepcludex and the Structural Cost of Chronic HDV Control
Hepcludex / bulevirtide is a real clinical breakthrough: it is the first FDA-approved treatment for chronic hepatitis delta virus infection in the United States. But the approval does not only solve a therapeutic absence. It also creates a new institutional problem around chronic cost, treatment continuity, payer restriction, and unequal access.
Bulevirtide controls HDV by blocking viral entry into hepatocytes through NTCP receptor inhibition. It does not directly eradicate infected hepatocytes, and long-term data show that many patients lose response after treatment discontinuation. This places Hepcludex closer to a chronic orphan antiviral control model than to a finite-course curative antiviral.
The central question is not whether Hepcludex works. It does. The deeper question is whether healthcare systems can sustain a high-cost, non-curative antiviral for a globally distributed disease without turning access into a function of geography, reimbursement power, and private wealth.

FDA’s New Food Chemical Review Program and the New Burden of Safety
FDA’s reassessment of BHT and ADA is not the main story. The deeper shift is that food chemical safety is becoming a post-market accountability system.
For decades, many food chemicals were accepted through toxicological inference, exposure modeling, safety factors, historical use, GRAS determinations, and expert consensus. They were not usually validated through long-term human clinical testing comparable to pharmaceuticals.
The new FDA framework changes the burden of defense. Historical authorization is becoming less sufficient as a stand-alone argument. Companies may now need to show that a substance remains defensible under current science, current exposure patterns, vulnerable-population risk, and evolving regulatory expectations.
For manufacturers, ingredient suppliers, retailers, and investors, this creates a new category of risk: food chemical regulatory risk. The central issue is not fear. The central issue is defensibility.

Trump’s Executive Order, FDA National Priority Vouchers, and the New Strategic Framework for Psilocybin, Methylone, and Noribogaine
The FDA’s acceleration of psychedelic and psychedelic-adjacent therapies should not be mistaken for therapeutic validation or imminent broad commercialization. The National Priority Voucher may compress the regulatory review window, but it does not approve products, lower evidentiary standards, or resolve the clinical burdens surrounding safety, durability, abuse potential, trial integrity, and controlled delivery.
The selected programs — psilocybin for depression, methylone for PTSD, and noribogaine for alcohol use disorder — are moving under one policy framework, but they are not one pharmacological class. Each carries a different safety profile, regulatory failure mode, and market-access constraint.
The deeper institutional issue is timing. Regulatory acceleration may create strategic optionality for sponsors and investors, but it also exposes weak evidence faster. In a conflict-heavy geopolitical cycle, the veteran-health framing adds another layer: these therapies may become relevant not only to psychiatric innovation, but to broader readiness for trauma, addiction, and post-conflict mental-health burden.
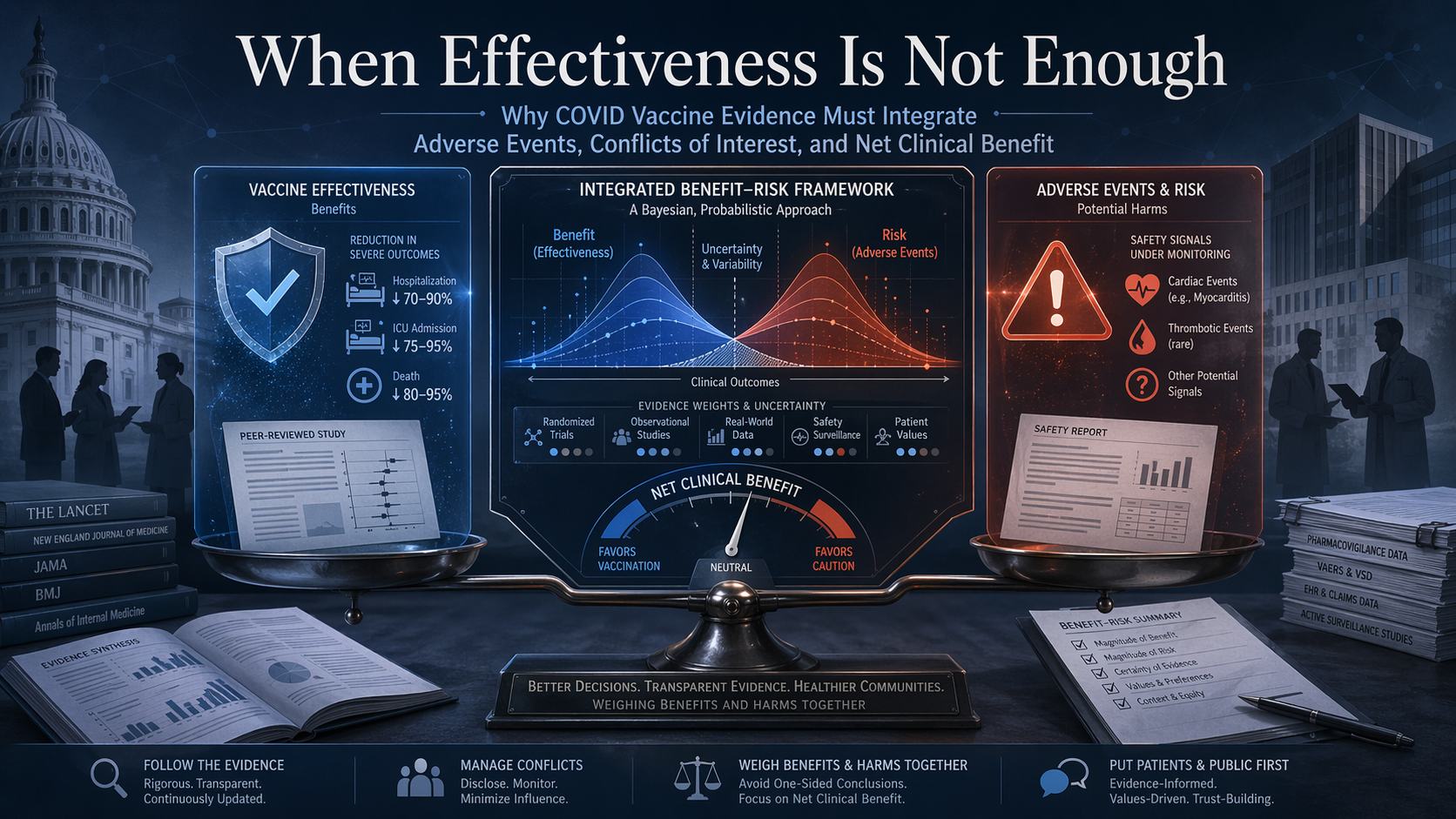
HHS’s Blocked CDC Report and the Limits of Effectiveness-Only Evidence
The main issue is no longer whether one blocked CDC report had methodological weaknesses. The deeper issue is that COVID vaccine evidence has often been communicated through a structurally incomplete model: effectiveness is measured in one channel, while adverse events and net benefit–risk balance remain outside the main frame. Two influential studies helped shape the public narrative of protection, but neither was designed to answer the full recommendation question. For preventive products, the real standard cannot be protection alone. It has to be whether expected benefit outweighs expected harm in the population being asked to receive the product.

Workshop on the Use of Bayesian Statistics in Clinical Development
The EMA’s Bayesian workshop makes one point increasingly clear: modern clinical development is searching for ways to preserve decision-making under conditions of evidence scarcity. Borrowing is one response, but it carries a structural risk—when external evidence is not truly transferable, efficiency can become distortion. The deeper regulatory question is therefore not only how to import evidence, but how to preserve its continuity, interpretability, and safety meaning across the full lifecycle.


Replimune’s CRL and the Cost of Weak Early-Phase Design
Replimune’s FDA Complete Response Letter should not be read as a routine regulatory delay. It exposed a deeper structural problem: when early-phase studies are built to generate signal rather than establish clean causal evidence, the weakness does not remain confined to the clinic. It compounds across the program, undermining approval, compressing investor confidence, and destabilizing the company’s operating model. Under a more explicit Bayesian FDA, that vulnerability becomes even harder to manage, because ambiguous contribution of effect, weak comparators, and fragile inferential logic are less likely to be absorbed and more likely to be challenged directly.

FDA’s New Endotoxins Guidance and the Shift from Release Testing to Lifecycle Quality Control
The FDA’s March 2026 endotoxins guidance should not be read as a narrow laboratory update. It signals a broader shift in regulatory expectations, moving endotoxin control from a release-test routine to a product-specific lifecycle quality strategy tied directly to Module 3, QA/QC, CMC, and patient safety. For firms, investors, and executive teams, the real issue is no longer whether a compliant procedure exists, but whether the company can defend its quality logic as a scientifically integrated system under a more demanding FDA standard.

NEJM’s HbF Promoter-Editing Papers, the Beam–Editas Split, and the Real Constraint on Gene-Edited Sickle Cell Therapy
Two recent New England Journal of Medicine papers have reinforced the therapeutic promise of HbF reactivation in sickle cell disease, but they also exposed a more important structural divide. The central question is no longer whether promoter editing can work. It is whether genomic elegance can survive the combined pressure of long-term safety uncertainty, multimillion-dollar treatment economics, and post-first-mover competition. Beam Therapeutics continues to advance its adenine base-editing strategy as a differentiated platform. Editas Medicine, despite publication-grade data, chose to discontinue reni-cel. The split reveals a harder truth now facing the field: in advanced gene editing, scientific validation alone is no longer enough.
The Logic Flow Behind FDA’s 2026 Shift: From Plausible Mechanism to NAM Validation
FDA’s March 18 NAM draft did not emerge in isolation. Read alongside the February Plausible Mechanism guidance, it points to a broader regulatory direction: away from rigid evidentiary defaults and toward more context-specific, human-relevant, and fit-for-purpose validation. BBIU’s earlier institutional reading had already identified that shift. The deeper implication is not weaker safety, but a higher burden of upstream verification—especially where preventable risk can be forced before irreversible patient exposure.

FDA AEMS and the Next Phase of Drug Safety Oversight
FDA’s new AEMS platform is not just a reporting upgrade. It is a meaningful shift toward integrated, real-time post-market safety surveillance across FDA-regulated products. But stronger visibility does not automatically produce stronger interpretability. While AEMS improves signal detection, transparency, and regulatory coordination, FDA is explicit that the system does not establish causality on its own. This analysis argues that the deeper regulatory frontier is not surveillance alone, but lifecycle safety continuity: a framework that links mechanistic risk prediction, clinical follow-up quality, unresolved-case review, and post-marketing therapeutic context into one auditable architecture.
Remibrutinib (LOU064): Structural Classification of a Multi-Indication Phase III Clinical Program
Remibrutinib (LOU064) is not being developed as a single-indication asset, nor as a speculative expansion play.
Its Phase III and Phase IIIb clinical architecture reveals a deliberate, multi-axis strategy aimed at establishing a long-term, orally administered BTK inhibitor across chronic inflammatory and neuro-immune diseases.
Starting from chronic urticaria as the anchor indication, Novartis has constructed a program that combines pivotal replicates, head-to-head comparator trials, randomized withdrawal designs, pediatric extensions, and event-driven studies in progressive disease. The result is a development profile focused not only on efficacy, but on durability, positioning, and lifecycle control.
This document provides a structured classification of the full Phase III remibrutinib program, mapping indications, trial design logic, and strategic intent. It also highlights the constraints that define the ceiling of expansion: chronic exposure, long-term safety, and regulatory scrutiny inherent to covalent BTK inhibition.
What emerges is a controlled expansion strategy—broad in scope, but tightly bounded by safety architecture—suggesting that remibrutinib is being positioned as a foundational immunomodulatory platform rather than a single commercial bet.